How to Use a Hex Editor to Read a Sam File
Contents
- one What is SAM
- 2 What Information is in SAM & BAM
- 2.1 What Information Does SAM/BAM Have for an Alignment
- 2.one.1 What is a CIGAR?
- 2.1.2 What is QUAL?
- 2.one.3 What are TAGs?
- two.2 What Data is in the SAM/BAM Header
- 2.1 What Information Does SAM/BAM Have for an Alignment
- 3 Instance SAM
- 3.ane Example Header Lines
- 3.2 Example Alignments
- four Tips/Tricks
What is SAM
The SAM Format is a text format for storing sequence data in a serial of tab delimited ASCII columns.
Most often it is generated as a human readable version of its sister BAM format, which stores the same data in a compressed, indexed, binary form.
Currently, virtually SAM format information is output from aligners that read FASTQ files and assign the sequences to a position with respect to a known reference genome. In the time to come, SAM will besides be used to archive unaligned sequence data generated directly from sequencing machines.
The current definition of the format is at [BAM/SAM Specification].
If yous are writing software to read SAM or BAM data, our C++ libStatGen is a good resource to use.
What Data is in SAM & BAM
SAM files and BAM files contain the same information, but in a different format. Refer to the specs to meet a format description.
Both SAM & BAM files incorporate an optional header section followed by the alignment section.
The header section may incorporate information nigh the entire file and additional data for alignments. The alignments then associate themselves with specific header information.
The alignment section contains the data for each sequence about where/how it aligns to the reference genome.
What Information Does SAM/BAM Have for an Alignment
Each Alignment has:
- query proper name, QNAME (SAM)/read_name (BAM). It is used to grouping/identify alignments that are together, like paired alignments or a read that appears in multiple alignments.
- a bitwise ready of data describing the alignment, FLAG. Provides the following data:
- are there multiple fragments?
- are all fragments properly aligned?
- is this fragment unmapped?
- is the adjacent fragment unmapped?
- is this query the reverse strand?
- is the adjacent fragment the reverse strand?
- is this the 1st fragment?
- is this the concluding fragment?
- is this a secondary alignment?
- did this read neglect quality controls?
- is this read a PCR or optical duplicate?
Not all alignments contain The residual of the alignment fields may be set to default values if the information is unknown.
- reference sequence proper name, RNAME, frequently contains the Chromosome proper name.
- leftmost position of where this alignment maps to the reference, POS. For SAM, the reference starts at ane, so this value is ane-based, while for BAM the reference starts at 0,so this value is 0-based. Beware to always apply the correct base of operations when referencing positions.
- mapping quality, MAPQ, which contains the "phred-scaled posterior probability that the mapping position" is wrong. (run across [[one]])
- cord indicating alignment information that allows the storing of clipped, CIGAR
- the reference sequence name of the next alignment in this group, MRNM or RNEXT. In paired alignments, it is the mate's reference sequence proper noun. (A grouping is alignments with the same query proper name.)
- leftmost position of where the next alignment in this grouping maps to the reference, MPOS or PNEXT. For SAM, the reference starts at 1, so this value is 1-based, while for BAM the reference starts at 0,and then this value is 0-based. Beware to always utilize the correct base when referencing positions.
- length of this group from the leftmost position to the rightmost position, ISIZE or TLEN
- the query sequence for this alignment, SEQ
- the query quality for this alignment, QUAL, i for each base in the query sequence.
- Boosted optional data is also contained within the alignment, TAGs. A bunch of different data tin be stored here and they appear every bit primal/value pairs. Come across the spec for a detailed list of commonly used tags and what they mean.
What is a CIGAR?
You may have heard the term CIGAR, but wondered what it ways. Hopefully this section will help clarify information technology.
The sequence existence aligned to a reference may have additional bases that are not in the reference or may be missing bases that are in the reference. The CIGAR string is a sequence of of base lengths and the associated performance. They are used to signal things similar which bases align (either a friction match/mismatch) with the reference, are deleted from the reference, and are insertions that are non in the reference.
For example:
RefPos: i 2 three four five 6 7 8 9 10 11 12 13 fourteen 15 16 17 18 xix Reference: C C A T A C T G A A C T G A C T A A C Read: ACTAGAATGGCT
Aligning these two:
RefPos: one 2 iii 4 5 six 7 viii 9 x xi 12 xiii xiv 15 xvi 17 18 19 Reference: C C A T A C T G A A C T 1000 A C T A A C Read: A C T A One thousand A A T Chiliad Thou C T
With the alignment above, yous get:
POS: v CIGAR: 3M1I3M1D5M
The POS indicates that the read aligns starting at position 5 on the reference. The CIGAR says that the first three bases in the read sequence align with the reference. The next base in the read does non exist in the reference. Then 3 bases align with the reference. The adjacent reference base does non exist in the read sequence, then five more bases marshal with the reference. Note that at position xiv, the base in the read is different than the reference, just information technology still counts every bit an 1000 since information technology aligns to that position.
What is QUAL?
QUAL stands for query quality. It is an indicator for how authentic each base in the query sequence (SEQ) is. If QUAL is specified, there is a quality value for each base in SEQ.
Quality is calculated based on the probability that a base is incorrect, p, using the following formula:
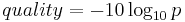
This quality is called the Phred Quality Score.
Since a human being readable format is desired for SAM, 33 is added to the calculated quality in order to make it a printable character ranging from ! - ~.
And so, for SAM, the QUAL field is:
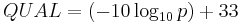
Phred Quality is besides plant in a FASTQ file, described hither: http://en.wikipedia.org/wiki/FASTQ_format#Quality
What are TAGs?
TAGs are optional fields on a SAM/BAM Alignment. A TAG is comprised of a two character TAG cardinal, they blazon of the value, and the value:
[A-Za-z][A-za-z]:[AifZH]:.*
The types, A, i, f, Z, H are used to bespeak the type of value stored in the tag.
| Type | Description |
|---|---|
| A | character |
| i | signed 32-bit integer |
| f | unmarried-precision float |
| Z | string |
| H | hex string |
There are a set of predefined tags that are full general used in Alignments. They are documented in the SAM Specification. Predefined tags accept been specified for storing information about the read or alignment. Examples of things stored in predefined tags:
- Previous settings for various fields if they have been updated due to additional processing
- Mappings from the alignment to Header values, used to match to a read grouping or program.
- Additional data which may already be in the header like library and platform.
A user can as well use any additional tags to store whatsoever information they desire. TAGs starting with 10, Y, or Z are reserved to exist user defined.
Examples:
XT:A:U - user divers tag called XT. It holds a character. The value associated with this tag is 'U'. NM:i:2 - predefined tag NM means: Edit distance to the reference (number of changes necessary to brand this equal the reference, excluding clipping)
The SAM/BAM header is non required, merely if it is in that location, it contains generic data for the SAM/BAM file.
The header may comprise the version information for the SAM/BAM file and information regarding whether or not and how the file is sorted.
It besides contains supplemental information for alignment records like information about the reference sequences, the processing that was used to generate the various reads in the file, and the programs that have been used to process the different reads. The alignment records may then indicate to this supplemental information identifying which ones the specific alignment is associated with.
For instance, a grouping of reads in the SAM/BAM file may all exist assigned to the same reference sequence. Rather than every alignment containing data about the reference sequence, this data is put in the header, and the alignment "points" to the appropriate reference sequence in the header via the RNAME field. The header contains generic information about this reference similar its length.
The SAM/BAM Header also may contain comments which are free-form text lines that can incorporate any data.
Header lines start with an '@'.
Example SAM
@HD VN:i.0 SO:coordinate @SQ SN:1 LN:249250621 As:NCBI37 UR:file:/information/local/ref/GATK/human_g1k_v37.fasta M5:1b22b98cdeb4a9304cb5d48026a85128 @SQ SN:two LN:243199373 AS:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta M5:a0d9851da00400dec1098a9255ac712e @SQ SN:3 LN:198022430 As:NCBI37 UR:file:/data/local/ref/GATK/human_g1k_v37.fasta M5:fdfd811849cc2fadebc929bb925902e5 @RG ID:UM0098:1 PL:ILLUMINA PU:HWUSI-EAS1707-615LHAAXX-L001 LB:lxxx DT:2010-05-05T20:00:00-0400 SM:SD37743 CN:UMCORE @RG ID:UM0098:2 PL:ILLUMINA PU:HWUSI-EAS1707-615LHAAXX-L002 LB:80 DT:2010-05-05T20:00:00-0400 SM:SD37743 CN:UMCORE @PG ID:bwa VN:0.five.4 @PG ID:GATK TableRecalibration VN:i.0.3471 CL:Covariates=[ReadGroupCovariate, QualityScoreCovariate, CycleCovariate, DinucCovariate, TileCovariate], default_read_group=null, default_platform=null, force_read_group=null, force_platform=null, solid_recal_mode=SET_Q_ZERO, window_size_nqs=v, homopolymer_nback=7, exception_if_no_tile=false, ignore_nocall_colorspace=imitation, pQ=5, maxQ=40, smoothing=one
In the alignment examples beneath, you will see that the 2nd alignment maps back to the RG line with ID UM0098.1, and all of the alignments bespeak back to the SQ line with SN:one because their RNAME is 1.
Case Alignments
This is what the alignment section of a SAM file looks like:
1:497:R:-272+13M17D24M 113 1 497 37 37M 15 100338662 0 CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==nine;>>>>>=>>>>>>>>>>>=>>>>>>>>>> XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:i X1:i:0 XM:i:0 XO:i:0 XG:i:0 Doc:Z:37 19:20389:F:275+18M2D19M 99 1 17644 0 37M = 17919 314 TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>iv::>>:<ix RG:Z:UM0098:i XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:four X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 19:20389:F:275+18M2D19M 147 1 17919 0 18M2D19M = 17644 -314 GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<eight<8<<<8<<><<<<><seven<;<<<>><< XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:1 XG:i:two MD:Z:18^CA19 9:21597+10M2I25M:R:-209 83 ane 21678 0 8M2I27M = 21469 -244 CACCACATCACATATACCAAGCCTGGCTGTGTCTTCT <;9<<5><<<<><<<>><<><>><9>><>>>nine>>><> XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:5 X1:i:0 XM:i:0 XO:i:i XG:i:two MD:Z:35
In this instance, the fields are:
| Field | Alignment one | Alignment ii | Alignment 3 | Alignment iv |
|---|---|---|---|---|
| QNAME | i:497:R:-272+13M17D24M | 19:20389:F:275+18M2D19M | nineteen:20389:F:275+18M2D19M | 9:21597+10M2I25M:R:-209 |
| FLAG | 113 | 99 | 147 | 83 |
| RNAME | one | 1 | 1 | 1 |
| POS | 497 | 17644 | 17919 | 21678 |
| MAPQ | 37 | 0 | 0 | 0 |
| CIGAR | 37M | 37M | 18M2D19M | 8M2I27M |
| MRNM/RNEXT | 15 | = | = | = |
| MPOS/PNEXT | 100338662 | 17919 | 17644 | 21469 |
| ISIZE/TLEN | 0 | 314 | ||
| SEQ | CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG | TATGACTGCTAATAATACCTACACATGTTAGAACCAT | GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT | CACCACATCACATATACCAAGCCTGGCTGTGTCTTCT |
| QUAL | 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> | >>>>>>>>>>>>>>>>>>>><<>>><<>>four::>>:<9 | ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< | <;ix<<v><<<<><<<>><<><>><ix>><>>>9>>><> |
| TAGs | XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 Md:Z:37 | RG:Z:UM0098:1 XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:iv X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37 | XT:A:R NM:i:ii SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:eighteen^CA19 | XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:5 X1:i:0 XM:i:0 XO:i:1 XG:i:two Md:Z:35 |
Tips/Tricks
- Calculating BAM Block Size
- Block Size = eight*iv + ReadNameLength(including null) + CigarLength*four + (ReadLength+1)/ii + ReadLength + TagLength
Y'all should now be a SAM expert :-)
Source: https://genome.sph.umich.edu/wiki/SAM
{kind=link}
Post a Comment for "How to Use a Hex Editor to Read a Sam File"